
Hello World! What’s a conformer?

Hi there!
My name is Nate and I’m a data scientist in the biotech world. I’ve been wanting to start a blog as a “how to” resource for working with chemistry in Python. This is my first post (so please be kind) but I’d love feedback in the comments. I also welcome ideas for new topics!
To start this blog, I wanted to make my first post about something near and dear to my heart conformers.
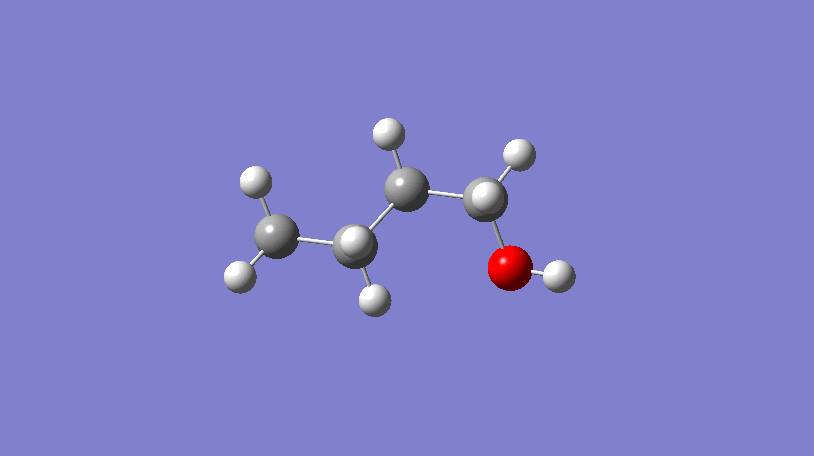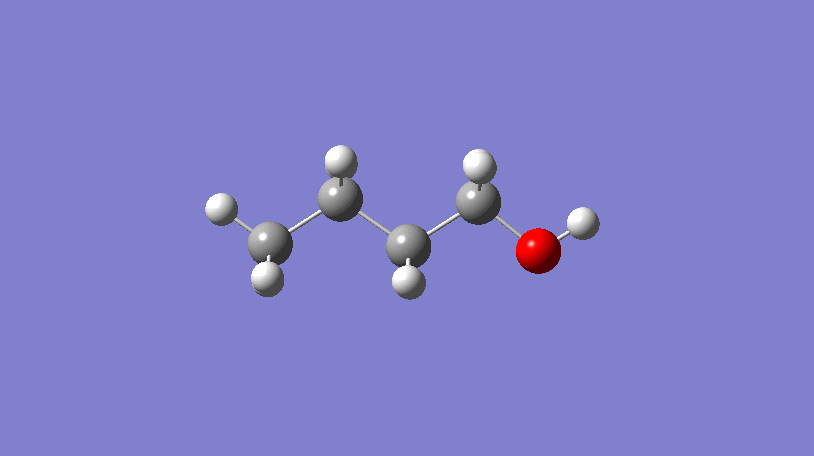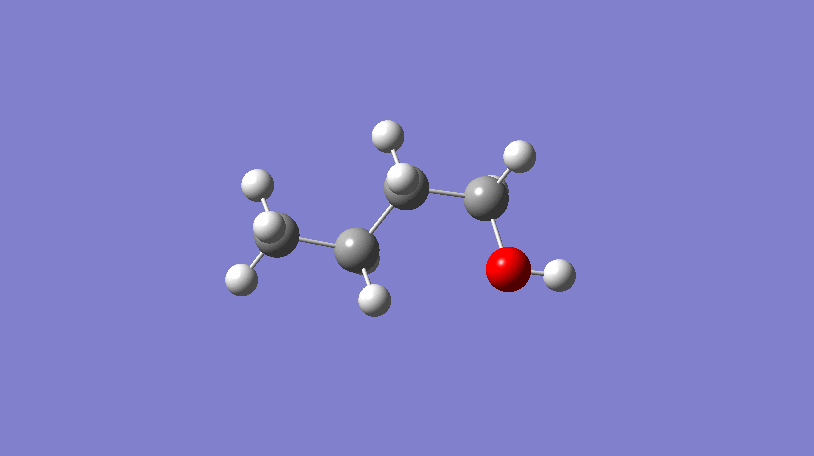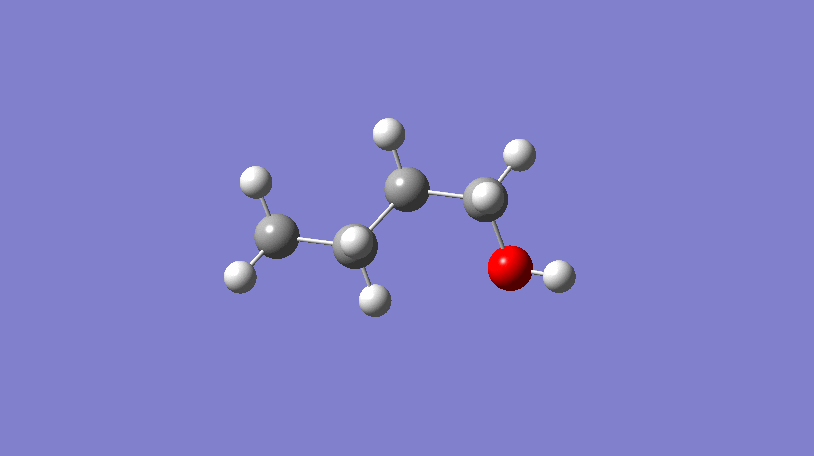
In the video above, a 3D representation of n-butanol (CCCCO) undergoes a conformational change by rotating about a C-C bond, ending in a final conformer.
In short, conformers are what describe the 3D geometry of a molecule and can be very useful!
Many properties can be calculated when you have a reasonable geometry of a molecule. This includes (but is not limited to)…
enthalpy & entropy using statistical mechanics and thermodynamic relations (e.g.)
ligand-protein interactions using docking simulations and molecular dynamics
High-stakes R&D decisions are often based on these calculated molecular properties, so the calculations ought to be accurate — and accuracy starts with the conformer’s geometry. Each conformer has a geometry-dependant energetic state and these states directly impact all of these properties. By finding the most probable geometry (or set of geometries), you can more accurately calculate properties that closely match one’s experimental conditions.
The question is, how do we generate conformers with a computer?
This is where RDKit comes in
(you’re going to be hearing about RDKit a lot in most of these blog posts)
RDKit is an industry-standard open-source cheminformatic software
compiled in C++ with an easy-to-use Python wrapper

This is example code to create virtual molecules using RDKit
You use string-based representations, called SMILES strings to create virtual molecules. The molecules can be treated as a chemical graph with atoms as nodes, which are connected by bonds as edges.
RDKit also has a general understanding of the “allowable distances” between atoms. It uses these distances to construct a bounds matrix when creating the 3D geometry.
The bounds matrix (BM) is an N x N matrix, where N is the number of atoms. Each entry in the BM represents an allowable distance between atoms (i.e. upper and lower bounds) with “0” along the diagonal. When creating the 3D geometry, atoms are placed in a square box of length, l = 2 * Max(BM) and the distances between atoms are measured to create an N x N distance matrix. If all values in the distance matrix are not within the BM limits, the algorithm will try again. It will repeat this process until it finds a geometry that’s within the BM limits, or the maximum number of iterations has been reached.
The resultant geometry can be visualized, modified, and saved before it is used to calculate other properties.
So, let’s search the internet and figure out how to do it ourselves!
What would ChatGPT do?
〰️
What would ChatGPT do? 〰️
To start from nothing, I asked ChatGPT the following question:
How can I generate multiple low energy conformers using RDKit?
And I got back the following response:
RDKit is a powerful cheminformatics toolkit that includes functionality for generating conformers of molecules. Conformers are different spatial arrangements of atoms in a molecule that represent different low-energy structures the molecule can adopt. Here's a basic outline of how you can generate conformers using RDKit in Python:
- Install RDKit: If you haven't already, you'll need to install RDKit. You can typically do this using Python package managers like pip or conda.
pip install rdkit
- Import necessary modules: In your Python script, import the necessary RDKit modules:
from rdkit import Chem
from rdkit.Chem import AllChem
- Load your molecule: You need to create an RDKit molecule object from your molecule's SMILES string or from an SDF file:
# Load molecule from SMILES string
smiles = "CCO" # Example SMILES string
mol = Chem.MolFromSmiles(smiles)
- Generate conformers: Use the
AllChem.EmbedMultipleConfs()function to generate conformers. You can specify parameters such as the number of conformers to generate and the maximum number of iterations for each conformer generation:
num_conformers = 10
AllChem.EmbedMultipleConfs(mol, numConfs=num_conformers)
- Optimize conformers: Optionally, you can perform energy minimization to optimize the conformers:
for conf in mol.GetConformers():
AllChem.UFFOptimizeMolecule(mol, confId=conf.GetId())
- Access conformers: You can access the generated conformers from the molecule object and extract information such as their coordinates:
for i, conf in enumerate(mol.GetConformers()):
print(f"Conformer {i+1} coordinates:")
for atom in conf.GetPositions():
print(atom)
This is a basic example to get you started with generating conformers using RDKit. Depending on your specific requirements, you may need to adjust parameters or add additional steps for your application. RDKit offers a wide range of functionality for working with molecular structures, so it's worth exploring the documentation for more advanced usage.
In all honesty, ChatGPT got it right…
… but I’d correct a few things. First off, AllChem.UFFOptimizeMolecule runs on a single thread making this iterative approach pretty slow. You could use AllChem.UFFOptimizeMoleculeConfs(..., numThreads=0) instead — this will use all available threads and will speed up the workflow. Also, ChatGPT’s answer didn’t include details about finding the low-energy conformers or about viewing the geometries.
Don’t get me wrong, it’s a great response —
but I think a human can do better.
I put together a Colab Notebook with much of ChatGPT’s suggestions, but I built upon it further — click the button to see the code!
The notebook will start by showing you how to use hi-chem, the Python package that I’ll be releasing code to. Then it will break down how to perform the conformer analysis, piece by piece. I included screenshots of the important parts of the code — this outlines how to parse the energies from the conformers and how to view multiple conformers together using py3Dmol.




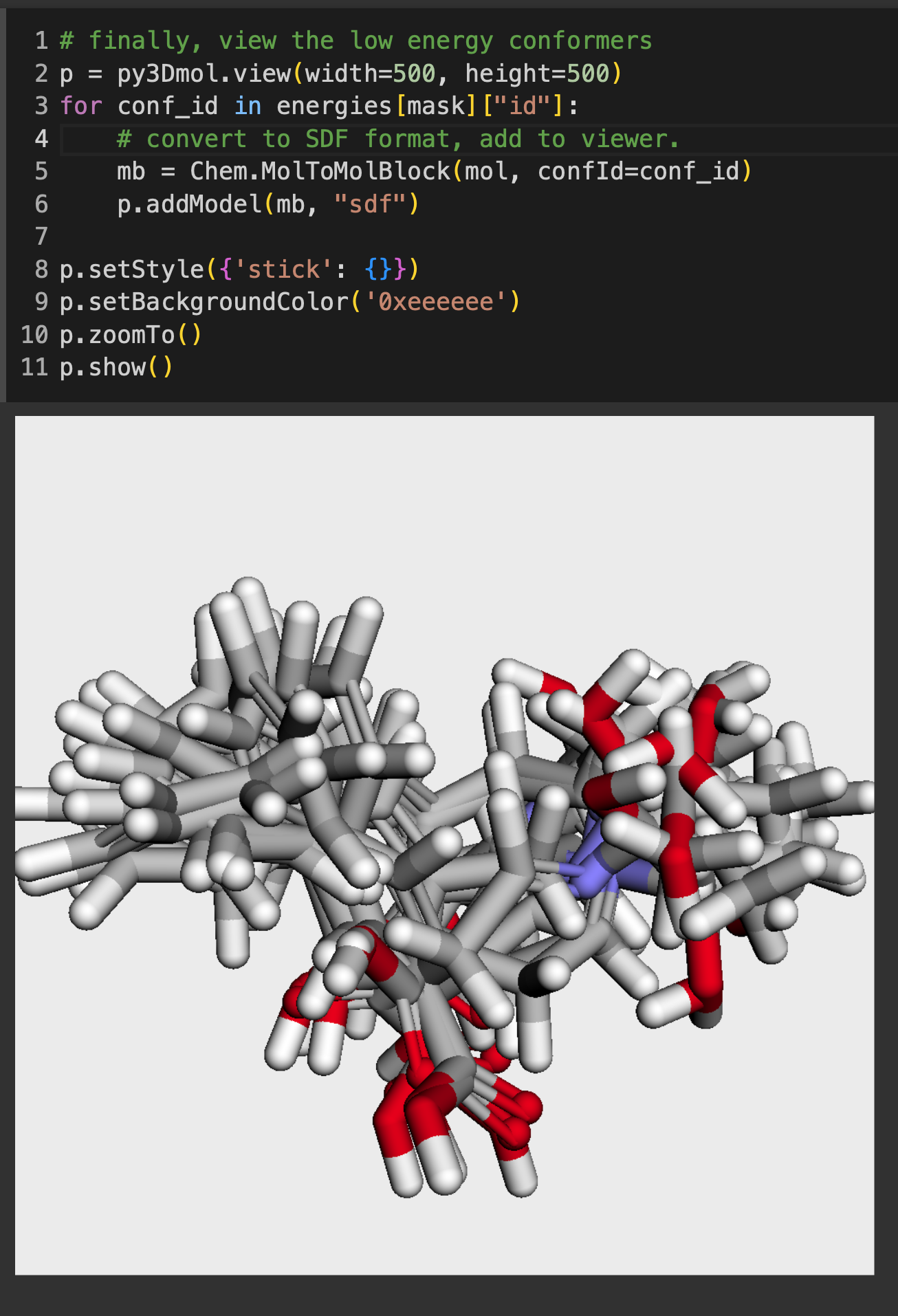
And again, ChatGPT got us part of the way there — we were able to generate conformers successfully but it was missing some functionality. So please, give this code a try the next time you need to generate conformers, and let me know what you think!
Thank you very much and I’m looking forward to making more posts 😎